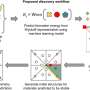
Breakthrough algorithm expands the exploration space for materials by orders of magnitude

Nanoengineers at the University of California San Diego's Jacobs School of Engineering have developed an AI algorithm that predicts the structure and dynamic properties of any material—whether existing or new—almost instantaneously. Known as M3GNet, the algorithm was used to develop matterverse.ai, a database of more than 31 million yet-to-be-synthesized materials with properties predicted by machine learning algorithms. Matterverse.ai facilitates the discovery of new technological materials with exceptional properties.
The team behind M3GNet, led by UC San Diego nanoengineering professor Shyue Ping Ong, uses matterverse.ai and the new capabilities of M3GNet in their search for safer and more energy-dense electrodes and electrolytes for rechargeable lithium-ion batteries. The project is explored in the Nov. 28 issue of the journal Nature Computational Science.
The properties of a material are determined by the arrangement of its atoms. However, existing approaches to obtain that arrangement are either prohibitively expensive or ineffective for many elements.
"Similar to proteins, we need to know the structure of a material to predict its properties." said Ong, the associate director of the Sustainable Power and Energy Center at the Jacobs School of Engineering. "What we need is an AlphaFold for materials."
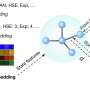
AlphaFold is an AI algorithm developed by Google DeepMind to predict protein structure. To build the equivalent for materials, Ong and his team combined graph neural networks with many-body interactions to build a deep learning architecture that works universally, with high accuracy, across all the elements of the periodic table.
"Mathematical graphs are really natural representations of a collection of atoms," said Chi Chen, a former senior project scientist in Ong's lab and first author of the work, who is now a senior quantum architect at Microsoft Quantum. "Using graphs, we can represent the full complexity of materials without being subject to the combinatorial explosion of terms in traditional formalisms."
To train their model, the team used the huge database of materials energies, forces and stresses collected in the Materials Project over the past decade. The result is the M3GNet interatomic potential (IAP), which can predict the energies and forces in any collection of atoms. Matterverse.ai was generated through combinatorial elemental substitutions on more than 5,000 structural prototypes in the Inorganic Crystal Structure Database (ICSD). The M3GNet IAP was then used to obtain the equilibrium crystal structure—a process called "relaxation"—for property prediction.
Of the 31 million materials in matterverse.ai today, more than a million are predicted to be potentially stable. Ong and his team intend to greatly expand not just the number of materials, but also the number of ML-predicted properties, including high-value properties with small data sizes using a multi-fidelity approach they developed earlier.
Beyond structural relaxations, the M3GNet IAP also has broad applications in dynamic simulations of materials and property predictions as well.
"For instance, we are often interested in how fast lithium ions diffuse in a lithium-ion battery electrode or electrolyte. The faster the diffusion, the more quickly you can charge or discharge a battery," Ong said. "We have shown that the M3GNet IAP can be used to predict the lithium conductivity of a material with good accuracy. We truly believe that the M3GNet architecture is a transformative tool that can greatly expand our ability to explore new material chemistries and structures."
To promote the use of M3GNet, the team has released the framework as an open-source Python code on Github. Since posting the preprint on Arxiv in Feb 2022, the team has received interest from academic researchers and those in the industry. There are plans to integrate the M3GNet IAP as a tool in commercial materials simulation packages.
This work was authored by Chi Chen and Shyue Ping Ong at UC San Diego.
More information: Chi Chen, A universal graph deep learning interatomic potential for the periodic table, Nature Computational Science (2022). DOI: 10.1038/s43588-022-00349-3. www.nature.com/articles/s43588-022-00349-3