Credit: CC0 Public Domain
Scientists seek stable, high-energy batteries designed for the electric grid.
Bringing new sources of renewable energy like wind and solar power onto the electric grid will require specially designed large batteries that can charge when the sun is shining and give energy at night. One type of battery is especially promising for this purpose: The flow battery. Flow batteries contain two tanks of electrically active chemicals that exchange charge and can have large volumes that hold a lot of energy.
For researchers working on flow batteries, their chief concern involves finding target molecules that offer the ability to both store a lot of energy and remain stable for long periods of time.
To find the right flow battery molecules, researchers at the U.S. Department of Energy's (DOE) Argonne National Laboratory have turned to the power of artificial intelligence (AI) to search through a vast chemical space of over a million molecules. Discovering the right molecules requires optimizing between several different characteristics. "In these batteries, we know that a majority of the molecules that we need will have to satisfy multiple properties," said Argonne chemist Rajeev Assary. "By optimizing several properties simultaneously, we have a better shot of finding the best possible chemistry for our battery."
"Nature is never perfect; no single molecule is ideal in every way. Our model allows us to juggle different parameters to find the best fit," says Argonne chemist Rajeev Assary.
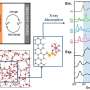
In a new study that follows on from work done last year, Assary and his colleagues in Argonne's Joint Center for Energy Storage Research modeled anolyte redoxmers, or electrically active molecules in a flow battery. For each redoxmer, the researchers identified three properties that they wanted to optimize. The first two, reduction potential and solvation free energy, relate to how much energy the molecule can store. The third, fluorescence, serves as a kind of self-reporting marker that indicates the overall health of the battery.
Because it is extraordinarily time consuming to calculate the properties of interest for all potential candidates, the researchers turned to a machine learning and AI technique called active learning, in which a model can actually train itself to identify increasingly plausible targets. "We're essentially looking for needles in haystacks," said Argonne postdoctoral researcher Hieu Doan. "When our model finds something that looks like a needle, it teaches itself how to find more."
For the most efficient use of active learning, the researchers started with a fairly small "haystack"—a dataset of 1400 redoxmer candidates whose properties they already knew from quantum mechanical simulations. By using this dataset as practice, they were able to see that the algorithm correctly identified the molecules with the best properties.
"In our previous research, we showed how we could optimize one property at a time, but trying to do several at once is a different kind of challenge and one that is probably more valuable for real-world conditions," Assary said. "Nature is never perfect; no single molecule is ideal in every way. Our model allows us to juggle different parameters to find the best fit."
Once they had explored the 1400-candidate set, the researchers expanded their search to a chemical space of a million different candidates. Through the model's iterative performance improvement, better and better molecules began to be identified. "We were encouraged by the fact that by looking at only 100 molecules, our model was already regularly finding molecules that had properties more attractive than those in our original dataset," Doan said.
According to Assary, the optimization algorithm could have uses beyond flow batteries. Conceivably, he said, this algorithm could be applied to other types of batteries and even other fields. "The mathematical approach we're using is also widely used by stock traders and data scientists, which goes to show how common optimization problems are," he said.
A paper based on the study, "Discovery of energy storage molecular materials using quantum chemistry-guided multiobjective Bayesian optimization," appeared in the October 14 issue of Chemistry of Materials.
In addition to Assary and Doan, other authors of the study include Argonne's Lily Robertson and Lu Zhang. Garvit Agarwal, formerly of Argonne but currently a scientist at Schrodinger, also contributed to the work.
More information: Garvit Agarwal et al, Discovery of Energy Storage Molecular Materials Using Quantum Chemistry-Guided Multiobjective Bayesian Optimization, Chemistry of Materials (2021). DOI: 10.1021/acs.chemmater.1c02040
Journal information: Chemistry of Materials
Provided by Argonne National Laboratory