Neural network that securely finds potential drugs could encourage large-scale pooling of sensitive data

MIT researchers have developed a cryptographic system that could help neural networks identify promising drug candidates in massive pharmacological datasets, while keeping the data private. Secure computation done at such a massive scale could enable broad pooling of sensitive pharmacological data for predictive drug discovery.
Datasets of drug-target interactions (DTI), which show whether candidate compounds act on target proteins, are critical in helping researchers develop new medications. Models can be trained to crunch datasets of known DTIs and then, using that information, find novel drug candidates.
In recent years, pharmaceutical firms, universities, and other entities have become open to pooling pharmacological data into larger databases that can greatly improve training of these models. Due to intellectual property matters and other privacy concerns, however, these datasets remain limited in scope. Cryptography methods to secure the data are so computationally intensive they don't scale well to datasets beyond, say, tens of thousands of DTIs, which is relatively small.
In a paper published in Science, researchers from MIT's Computer Science and Artificial Intelligence Laboratory (CSAIL) describe a neural network securely trained and tested on a dataset of more than a million DTIs. The network leverages modern cryptographic tools and optimization techniques to keep the input data private, while running quickly and efficiently at scale.
The team's experiments show the network performs faster and more accurately than existing approaches; it can process massive datasets in days, whereas other cryptographic frameworks would take months. Moreover, the network identified several novel interactions, including one between the leukemia drug imatinib and an enzyme ErbB4—mutations of which have been associated with cancer—which could have clinical significance.
"People realize they need to pool their data to greatly accelerate the drug discovery process and enable us, together, to make scientific advances in solving important human diseases, such as cancer or diabetes. But they don't have good ways of doing it," says corresponding author Bonnie Berger, the Simons Professor of Mathematics and a principal investigator at CSAIL. "With this work, we provide a way for these entities to efficiently pool and analyze their data at a very large scale."
Joining Berger on the paper are co-first authors Brian Hie and Hyunghoon Cho, both graduate students in electrical engineering and computer science and researchers in CSAIL's Computation and Biology group.
"Secret sharing" data
The new paper builds on previous work by the researchers in protecting patient confidentiality in genomic studies, which find links between particular genetic variants and incidence of disease. That genomic data could potentially reveal personal information, so patients can be reluctant to enroll in the studies. In that work, Berger, Cho, and a former Stanford University Ph.D. student developed a protocol based on a cryptography framework called "secret sharing," which securely and efficiently analyzes datasets of a million genomes. In contrast, existing proposals could handle only a few thousand genomes.
Secret sharing is used in multiparty computation, where sensitive data is divided into separate "shares" among multiple servers. Throughout computation, each party will always have only its share of the data, which appears fully random. Collectively, however, the servers can still communicate and perform useful operations on the underlying private data. At the end of the computation, when a result is needed, the parties combine their shares to reveal the result.
"We used our previous work as a basis to apply secret sharing to the problem of pharmacological collaboration, but it didn't work right off the shelf," Berger says.
A key innovation was reducing the computation needed in training and testing. Existing predictive drug-discovery models represent the chemical and protein structures of DTIs as graphs or matrices. These approaches, however, scale quadratically, or squared, with the number of DTIs in the dataset. Basically, processing these representations becomes extremely computationally intensive as the size of the dataset grows. "While that may be fine for working with the raw data, if you try that in secure computation, it's infeasible," Hie says.
The researchers instead trained a neural network that relies on linear calculations, which scale far more efficiently with the data. "We absolutely needed scalability, because we're trying to provide a way to pool data together [into] much larger datasets," Cho says.
The researchers trained a neural network on the STITCH dataset, which has 1.5 million DTIs, making it the largest publicly available dataset of its kind. In training, the network encodes each drug compound and protein structure as a simple vector representation. This essentially condenses the complicated structures as 1s and 0s that a computer can easily process. From those vectors, the network then learns the patterns of interactions and noninteractions. Fed new pairs of compounds and protein structures, the network then predicts if they'll interact.
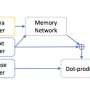
The network also has an architecture optimized for efficiency and security. Each layer of a neural network requires some activation function that determines how to send the information to the next layer. In their network, the researchers used an efficient activation function called a rectified linear unit (ReLU). This function requires only a single, secure numerical comparison of an interaction to determine whether to send (1) or not send (0) the data to the next layer, while also never revealing anything about the actual data. This operation can be more efficient in secure computation compared to more complex functions, so it reduces computational burden while ensuring data privacy.
"The reason that's important is we want to do this within the secret sharing framework ... and we don't want to ramp up the computational overhead," Berger says. In the end, "no parameters of the model are revealed and all input data—the drugs, targets, and interactions—are kept private."
Finding interactions
The researchers pitted their network against several state-of-the-art, plaintext (unencrypted) models on a portion of known DTIs from DrugBank, a popular dataset containing about 2,000 DTIs. In addition to keeping the data private, the researchers' network outperformed all of the models in prediction accuracy. Only two baseline models could reasonably scale to the STITCH dataset, and the researchers' model achieved nearly double the accuracy of those models.
The researchers also tested drug-target pairs with no listed interactions in STITCH, and found several clinically established drug interactions that weren't listed in the database but should be. In the paper, the researchers list the top strongest predictions, including: droloxifene and an estrogen receptor, which reached phase III clinical trials as a treatment for breast cancer; and seocalcitol and a vitamin D receptor to treat other cancers. Cho and Hie independently validated the highest-scoring novel interactions via contract research organizations.
Next, the researchers are working with partners to establish their collaborative pipeline in a real-world setting. "We are interested in putting together an environment for secure computation, so we can run our secure protocol with real data," Cho says.
More information: B. Hie el al., "Realizing private and practical pharmacological collaboration," Science (2018). science.sciencemag.org/cgi/doi … 1126/science.aat4807
This story is republished courtesy of MIT News (web.mit.edu/newsoffice/), a popular site that covers news about MIT research, innovation and teaching.