New machine learning approach speeds up search for molecular conformers

Conformer search continues to be a topic of great interest in computational chemistry, drug design and material science. It is a challenging endeavor due to the high dimensionality of the search space and the computational cost of accurate quantum chemical methods needed to determine the molecular structure and energy. Previously, searching for molecular conformers meant that thousands of structures needed to be relaxed first. Therefore, this process took up considerable time and computational resources even when applied to small molecules.
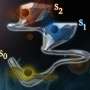
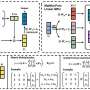
A recent paper authored by Lincan Fang, Esko Makkonen, Milica Todorovic, Patrick Rinke, and Xi Chen proposes a molecular conformer search procedure that combines an active learning Bayesian optimization (BO) algorithm with quantum chemistry methods to address this challenge. The BO active learning smartly samples the structures with low energies or high energy uncertainties, thus minimizing the required data points.
In this paper, the authors tested the procedure on four amino acids (cysteine, serine, tryptophan and aspartic acid). After only 1000 single-point calculations and approximately 80 structure relaxations, which is less than 10% of the computational cost of the current fastest method, the team found the low-energy conformers in good agreement with experimental measurements and reference calculations.
First author Fang now plans to extend the method to search for structures of molecules that are bonded to nanoclusters.
This research paper is published in the Journal of Chemical Theory and Computation and has been selected as a supplementary cover of the issue.
More information: Lincan Fang et al. Efficient Amino Acid Conformer Search with Bayesian Optimization, Journal of Chemical Theory and Computation (2021). DOI: 10.1021/acs.jctc.0c00648