November 9, 2023 feature
This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
A machine learning–based tool to model phase-change memory materials
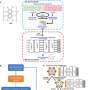
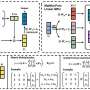
 contains crystalline structures, including Materials Project (MP) entries , and AIMD snapshots of liquid and amorphous phases. A two-step iterative training process is then carried out, with standard iterations (iter-1) and domain-specific iterations (iter-2) gradually expanding the database. b, Two-dimensional structural map representing the reference database, generated using the SOAP similarity matrix. Credit: Nature Electronics (2023). DOI: 10.1038/s41928-023-01030-x")
Computer simulations can greatly contribute to the study of new promising materials for technological applications. These include so-called phase-change materials (PCMs), substances that release or absorb thermal energy while melting and solidifying, which are promising for the development of memory components.
Researchers at University of Oxford and Xi'an Jiaotong University in China recently developed a machine learning model that could help to simulate these materials at an atomic scale, realistically replicating the conditions under which devices operate. Their model, presented in a paper published in Nature Electronics, can produce detailed simulations at a high speed, helping users to better understand the processes unfolding in devices based on PCMs.
"Our paper describes a computer simulation approach for modeling PCMs, which are used in digital data storage and processing," Volker Deringer, one of the researchers who carried out the study, told Tech Xplore. "Simulations of this type, called 'molecular dynamics' (MD), have typically been used to describe a few hundred atoms or so, and those small-scale simulations have already been very useful in the PCM community.
"We leverage the power of atomistic machine learning (ML) approaches to go much further, showing how we can reach the length scale of real devices, while still describing all individual atoms in the system with an accuracy comparable to that of quantum mechanics."
Deringer and his collaborators dubbed the simulations enabled by their model with the broad term device-scale atomistic modeling. This is because it is the first technique that can model more than half a million atoms inside memory devices under realistic device operation conditions.
The computations underpinning the machine learning model were performed by Yuxing Zhou, a final-year doctoral student who is part of Deringer's research lab in Oxford. While completing his Master's degree, Zhou had closely collaborated with Wei in Xi'an Jiaotong University and he also did so while conducting this study.
"We are not the first authors to describe an ML potential for PCMs—in fact, PCMs have been one of the early application cases for a popular class of neural-network potentials, with a foundational paper by Marco Bernasconi's group in Italy having been published already more than a decade ago," Deringer explained. "There is also work from colleagues in the U.K. on describing the ternary flagship compound, Ge2Sb2Te5."
Compared to other models introduced in past studies, the machine learning model developed by Zhou, Deringer and their collaborators was trained on a more diverse set of compounds, specifically all of the so-called quasi-binary line between GeTe and Sb2Te3. As a result, their model describes a wide range of materials that could interest researchers studying PCMs.
"The work provides an important proof-of-concept that a new type of simulation—at full device scale—is now possible for phase-change memories," Deringer said. "It shows applications of the ML potential to various, increasingly challenging scenarios: the growth of multiple crystalline grains (i.e., the 'digital ones'), the so-called non-isothermal heating of a large cell, and an example of an applied electric field—all these points are relevant to fully understanding the 'real-world' operation of PCM-based devices."
The researchers trained their model on a new dataset they compiled, which included labeled quantum mechanical data. After developing an initial version of the model, they gradually started feeding it data.
"The result of this iterative process is an ML-based interatomic potential—these types of potentials used to be quite specialized tools a few years ago, but have now become much more widely applied," Deringer said. "A key advance in the present paper is the development of the dataset that 'feeds' the ML model, which was carefully constructed to represent, for example, the atomic-scale structural changes during crystallization and growth of PCMs."
In initial tests, the model created by this team of researchers proved to be highly promising, enabling the detailed modeling of atoms in PCMs during multiple thermal cycles and while simulated devices were performing delicate operations. This demonstrates the feasibility of using machine learning to simulate entire devices based on PCMs at an atomic scale.
"We've taken great care to validate the model (which is particularly important for ML potentials), and we encourage others to try it as well—the parameter files and other data are provided openly to the community," Deringer said. "I hope that this work will help to further strengthen the important role of theory and simulation in the field of PCM research, and that it will eventually help to create more and more realistic models of these materials and the devices based on them."
The dataset and parameter files used by Zhou, Deringer and their colleagues are open-source and can be accessed on Zenodo, thus other teams could soon use them to train their machine learning techniques. In the future, their model could prove useful for material scientists and engineers worldwide, aiding the understanding and design of PCM-based memory devices.
"One key next step will be extending these ML models to describe other components relevant to memory devices, for example, ovonic threshold switching materials," Deringer added. "A long-term hope would be to provide a 'toolkit' of high-quality datasets and ML models that people in the community can use essentially off the shelf. Our current paper shows simulations on the length scale corresponding to real devices, but another outstanding challenge remains the time-scale barrier, thus we are also working hard to make our ML-driven simulations even faster."
More information: Yuxing Zhou et al, Device-scale atomistic modelling of phase-change memory materials, Nature Electronics (2023). DOI: 10.1038/s41928-023-01030-x
© 2023 Science X Network