Using artificial intelligence to advance energy technologies

Hongliang Xin, an associate professor of chemical engineering in the College of Engineering, and his collaborators have devised a new artificial intelligence framework that can accelerate discovery of materials for important technologies, such as fuel cells and carbon capture devices.
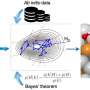
Titled "Infusing theory into deep learning for interpretable reactivity prediction," their paper in the journal Nature Communications details a new approach called TinNet—short for theory-infused neural network—that combines machine-learning algorithms and theories for identifying new catalysts. Catalysts are materials that trigger or speed up chemical reactions.
TinNet is based on deep learning, also known as a subfield of machine learning, which uses algorithms to mimic how human brains work. The 1996 victory of IBM's Deep Blue computer over world chess champion Garry Kasparov was one of the first advances in machine learning. More recently, deep learning has played a major role in the development of technologies such as self-driving cars.
Xin and his colleagues want to put machine learning to use in the field of catalysis for developing new and better energy technologies and products to improve daily life.
"About 90 percent of the products you see today are actually coming from catalysis," Xin said. The trick is finding the efficient and robust catalysts for each application, and finding new ones can be difficult.
"Understanding how catalysts interact with different intermediates and how to control their bond strengths to be in the Goldilocks Zone is absolutely the key to designing efficient catalytic processes," Xin said. "And our study provides a tool exactly for that."
Machine-learning algorithms can be helpful because they identify complex patterns in big data sets, something humans are not very good at, Xin said. But deep learning has limitations, especially when it comes to predicting highly complex chemical interactions—a necessary part of finding materials for a desired function. In these applications, sometimes deep learning fails, and it may not be clear why.
"Most of the machine-learning models developed for material properties prediction or classification are often considered 'black boxes' and provide limited physical insights," chemical engineering graduate student and paper co-author Hemanth Pillai said.
"The TinNet approach extends its prediction and interpretation capabilities, both of which are crucial in catalyst design." said Siwen Wang, also a chemical engineering graduate student and co-author of the study.
A hybrid approach, TinNet combines advanced theories of catalysis with artificial intelligence to help researchers peer into this "black box" of material design to understand what is happening and why, and it could help researchers break new ground in a number of fields.
"Hopefully we can make this approach generally accessible to the community and others can use the technique and really further develop the technique for renewable energy and decarbonization technologies that are crucial for the society," Xin said. "I think this is really the key technology that could make some breakthroughs."
Luke Achenie, a professor of chemical engineering specializing in machine learning, collaborated with Xin on the project, as well as graduate student Shih-Han Wang, who helped author the paper. Now the team is working on applying TinNet to their catalysis work. Andy Athawale, an undergraduate chemical engineering student, has joined the effort.
"I really love to see the different aspects of chemical engineering outside of the course of classes," Athawale said. "It has a lot of applications, and you know, it could be really revolutionary. So it's just amazing to be part of it."
More information: Shih-Han Wang et al, Infusing theory into deep learning for interpretable reactivity prediction, Nature Communications (2021). DOI: 10.1038/s41467-021-25639-8