September 15, 2022 feature
A molecular optimization framework to identify promising organic radicals for aqueous redox flow batteries

Recent advancements in the development of machine learning and optimization techniques have opened new and exciting possibilities for identifying suitable molecular designs, compounds, and chemical candidates for different applications. Optimization techniques, some of which are based on machine learning algorithms, are powerful tools that can be used to select optimal solutions for a given problem among a typically large set of possibilities.
Researchers at Colorado State University and the National Renewable Energy Laboratory have been applying state-of-the-art molecular optimization models to different real-world problems that entail identifying new and promising molecular designs. In their most recent study, featured in Nature Machine Intelligence, they specifically applied a newly developed, open-source optimization framework to the task of identifying viable organic radicals for aqueous redox flow batteries, energy devices that convert chemical energy into electricity.
"Our project was funded by an ARPA-E program that was looking to shorten how long it takes to develop new energy materials using machine learning techniques," Peter C. St. John, one of the researchers who carried out the study, told TechXplore. "Finding new candidates for redox flow batteries was an interesting extension of some of our previous work, including a paper published in Nature Communications and another in Scientific Data, both looking at organic radicals."
The new framework created by St. John and his colleagues was inspired by their previous work on molecular optimization. The framework essentially consists of the artificial intelligence (AI) tool AlphaZero, developed by DeepMind, coupled with a fast machine learning-derived model, made up of two graph neural networks trained on almost 100,000 quantum chemistry simulations.
The first of the graph neural networks was trained to predict oxidation and reduction potentials, two important parameters for determining how much energy can be stored in aqueous redox flow batteries. The second predicts the density of electrons and the local 3D environment, which have both been found to be associated with the lifetime of these batteries.
"We pose molecule optimization as a tree search, where we build molecules by adding components iteratively onto a growing structure," St. John explained. "The advantage of this approach is that we can prune off large branches of the search space where molecules start to show substructures that are unrealistic. We can therefore limit our search space to only molecules that meet a predetermined set of simple criteria."
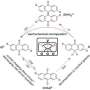
The researchers used their molecular optimization framework to run a series of tests aimed at identifying possible organic radicals for aqueous redox flow batteries that could be particularly stable and promising. The framework successfully identified several molecular candidates that satisfied a specific combination of criteria defined by St. John and his colleagues.
"We demonstrated that the set of possible candidates for a particular type of charge carrier in organic redox flow batteries may be larger than previously considered," St. John said. "We also showed that molecules may be found that could lead to simpler, high-performance batteries without requiring the use of transition metals."
So far, the optimization framework developed by this team of researchers has proved to be a highly promising tool for tackling complex real-world problems related to engineering and chemistry. In the future, it could thus be used to identify new desirable compounds and molecular candidates for many different technologies, including aqueous redox flow batteries.
"We would now like to explore adding additional criteria like solubility and redox pairs between charged states," St. John added. "This would require additional training data, but it may lead to more promising candidate structures."
More information: Shree Sowndarya S. V. et al, Multi-objective goal-directed optimization of de novo stable organic radicals for aqueous redox flow batteries, Nature Machine Intelligence (2022). DOI: 10.1038/s42256-022-00506-3
Peter C. St. John et al, Prediction of organic homolytic bond dissociation enthalpies at near chemical accuracy with sub-second computational cost, Nature Communications (2020). DOI: 10.1038/s41467-020-16201-z
Peter C. St. John et al, Quantum chemical calculations for over 200,000 organic radical species and 40,000 associated closed-shell molecules, Scientific Data (2020). DOI: 10.1038/s41597-020-00588-x
© 2022 Science X Network